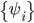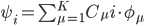Within the molecular orbital or Hartree-Fock theory, the multi-electron wavefunction is approximated by a single Slater determinant. where are molecular orbitals and are eigenfunctions of the Fock operator with the orbital energies as eigenvalues. The molecular orbitals are approximated as Linear Combination of Atomic Orbitals (LCAO) asand are delocalized over the entire molecule as shown in the case of ethylene below
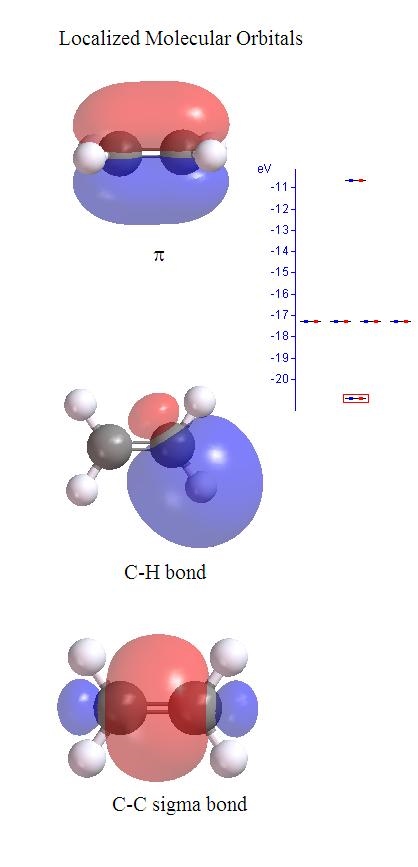
Orbtial energies shown in the figure above obey the Koopman's theorem and thus can be related to ionization potential and electron affinity of the molecule. Since the MO's are delocalized, they are more difficult to provide much physical interpretation. However, as a property of a determinant that a column can be added by another column multiplied by a constant without changing the value of the determinant. That means one can transform the delocalized MO into another set of MO and still does not change the total wavefunction and thus all observables of the molecule. There are several ways that one can transfer the delocalized MO's into localized MO's as shown below for ethylene.
In this case, the localized MO's show the nature of each bond. For instance, C=C has one sigma bond and one pi bond. Since localized MO's are not eigenfuctions of the Fock operator, orbital energies no longer have any physical meaning.
Exercise: Use Basic QChem Edu or Basic QChem tool with PsiViewer in Avisto to show the delocalized and localized MO's for BeH2. You can download these tools and Avisto from Astonis.